This semester, we explored how machine learning can accelerate drug discovery — specifically, how it can be used to predict molecular docking scores without running expensive simulations.
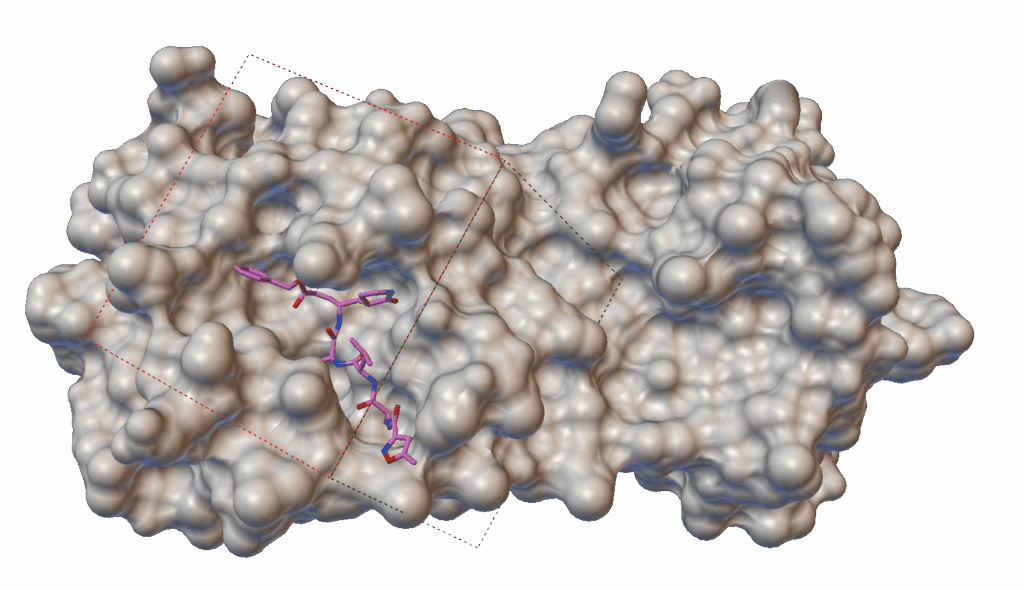
Docking simulations are commonly used to evaluate how well a small molecule (such as a drug candidate) binds to a target protein — in our case, the main protease (3CLpro) of SARS-CoV-2. These simulations produce a numerical docking score, which helps researchers prioritize promising compounds. However, running full simulations for tens of thousands of molecules can be extremely time-consuming.
To address this, we built a machine learning pipeline that predicts docking scores directly from a molecule’s structure.
From Molecule to Prediction
We started with molecular data in XYZ format, which contains the atomic numbers and 3D coordinates of each atom. This representation works well for simulations but isn’t suitable as input to a neural network. To solve this, we convert each molecule into a fixed-size numerical descriptor — a format that is invariant to translation, rotation, or the order of atoms.
We used the DScribe Python library to generate several types of descriptors:
- Coulomb Matrix (CM) – quick to compute, but sensitive to atom ordering.
- MBTR (Many-Body Tensor Representation) – captures up to 3-body interactions between atoms.
- SOAP (Smooth Overlap of Atomic Positions) – uses smooth density overlap and basis expansions to describe local environments.
- ACSF (Atom-Centered Symmetry Functions) – focuses on rotationally and translationally invariant local descriptors.
Each descriptor outputs either a matrix or a high-dimensional vector, which is used as input to a fully connected neural network trained to predict the docking score.
Pipeline Diagram
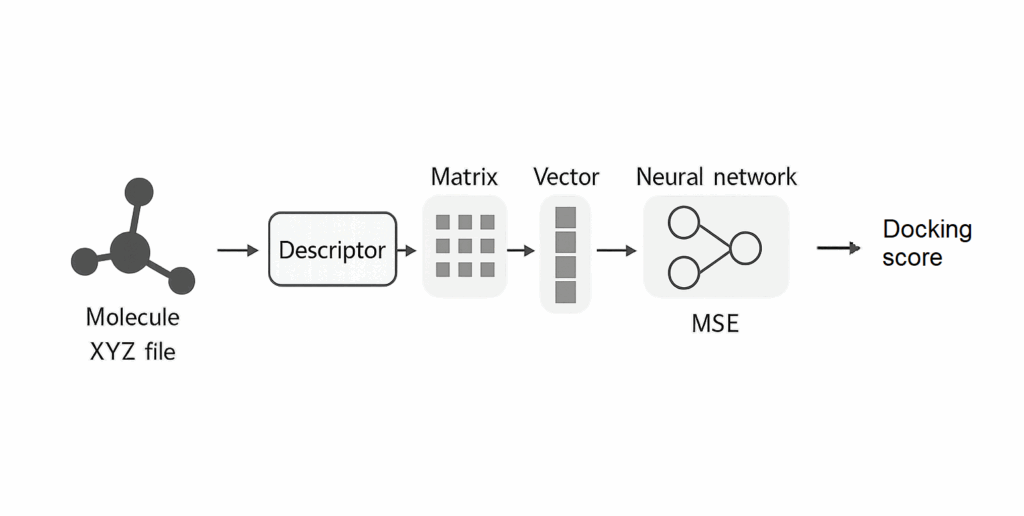
Here’s what happens at each stage:
- Molecule: Starts as a 3D structure in XYZ format.
- Descriptor: Converts the structure into a numerical format (e.g., CM, MBTR, SOAP, ACSF).
- Vectorization: Matrix descriptors are flattened into 1D vectors.
- Neural Network: The vector is passed to a regression model.
- Output: The model returns a predicted docking score, evaluated using Mean Squared Error (MSE).
Neural Network Setup
We implemented the regression model using TensorFlow. Each descriptor produces a unique input shape, but our model architecture remained relatively consistent:
- 3 hidden layers with 128, 64, and 32 neurons
- ReLU or Softplus activation functions
- Batch sizes of 32 to 128
- Optimizers: Adam or SGD
We used Hyperopt for hyperparameter tuning. Interestingly, our results showed that the choice and configuration of the descriptor had a much larger impact on performance than tweaks to the network architecture itself.
Results and Trade-offs
We trained and tested our model on a subset of the molecule dataset and evaluated it using MSE. Here’s a summary of how the different descriptors performed:
| Descriptor | Computation Time | MSE ↓ | Notes |
|---|---|---|---|
| CM | Very fast | ~0.63 | Poor accuracy, but quick |
| MBTR | Slow | ~0.34 | Captures detailed info, expensive to compute |
| SOAP | Moderate | ~0.29 | Best overall performance |
| ACSF | Medium | ~0.48 | Good accuracy, faster than SOAP |
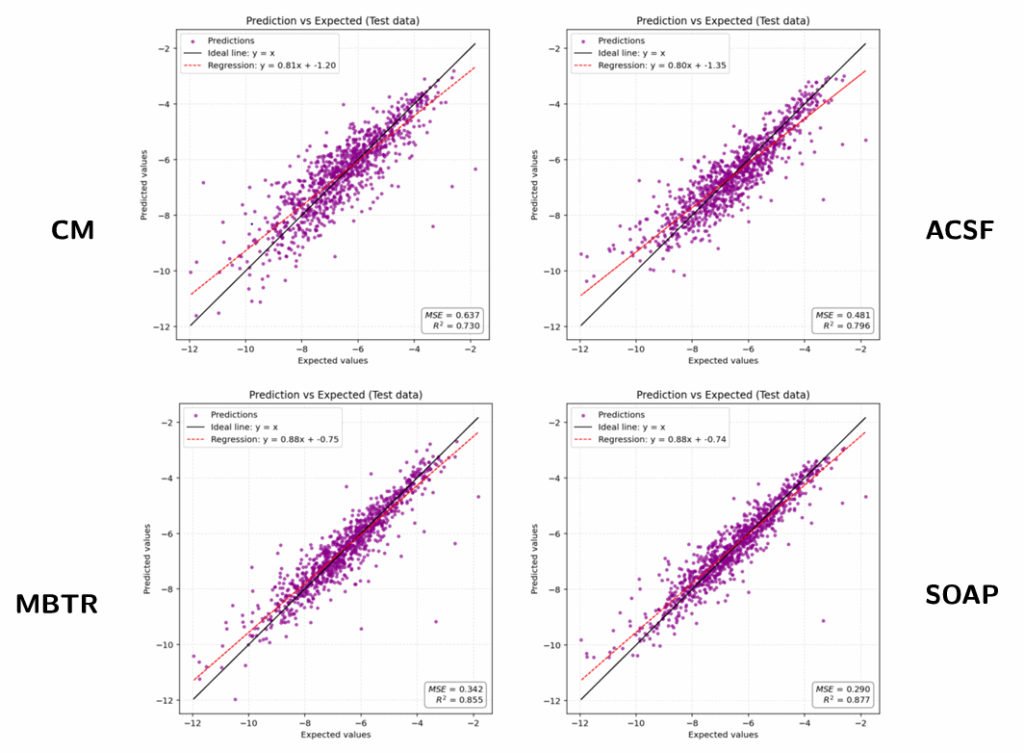
In general, SOAP and MBTR offered the best balance between accuracy and efficiency.
This project showed how deep learning can reduce the time needed for early-stage drug screening. Instead of computing docking scores from scratch, we can now make reliable predictions in a fraction of the time. It also highlighted the importance of choosing the right descriptor: the quality of the molecular input representation often matters more than the complexity of the neural network.